在這一章節,我們離開單純的「受體佔據」或「酵素抑制」。這裡的藥物(如 TKI 和免疫檢查點抑制劑)引發的是更為複雜的全身性反應——從微血管的退化、基因表現的改變,到免疫系統對內分泌腺體的無差別攻擊。
第一部分:Tyrosine Kinase Inhibitors (TKIs) 與血管內皮的崩解
代表藥物: Sunitinib, Sorafenib(用於腎細胞癌、甲狀腺癌等)。 這類藥物對甲狀腺的打擊是雙重的:既破壞結構,又消耗庫存。
1. 結構破壞:微血管退化 (Capillary Regression)
【微觀機制:為何甲狀腺首當其衝?】
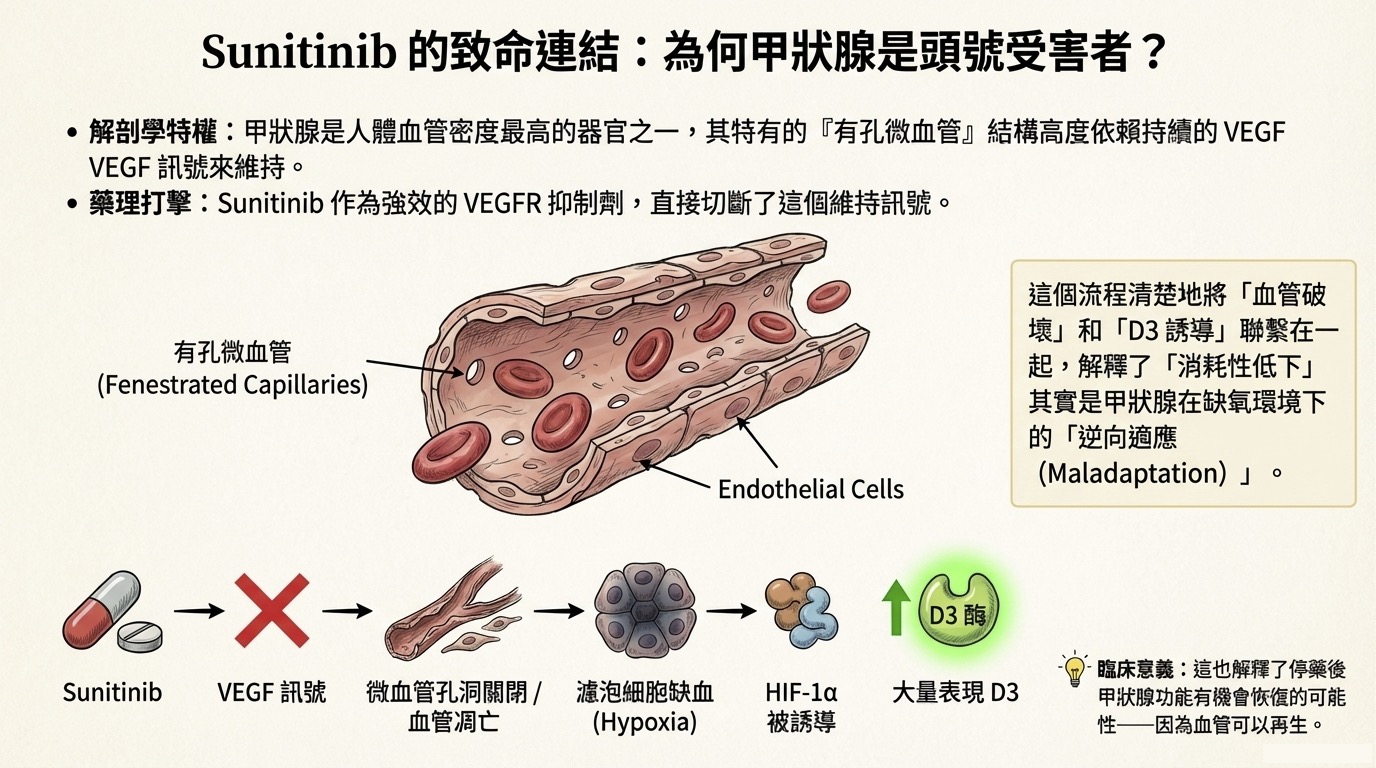
甲狀腺是人體血管密度最高的器官之一。為了快速交換荷爾蒙,其微血管具有特殊的 有孔結構 (Fenestrated Capillaries)。這些孔洞的維持,高度依賴持續的 VEGF (血管內皮生長因子) 訊號。
- 藥理打擊: Sunitinib 是強效的 VEGFR 抑制劑。阻斷訊號後,甲狀腺微血管的「孔洞」關閉,甚至導致微血管本身的凋亡與稀疏化 (Rarefaction)。
- 臨床進程:
- 缺血性壞死: 濾泡細胞因斷炊而壞死,引發破壞性甲狀腺炎 (Destructive thyroiditis)。
- 雙相變化: 初期因濾泡破裂,庫存荷爾蒙釋出導致短暫甲狀腺毒症 (Thyrotoxicosis);隨後因細胞死亡及血管退化,轉變為永久性的甲狀腺低下。
- 註:因血管具有再生能力,停藥後甲狀腺功能有機會恢復。
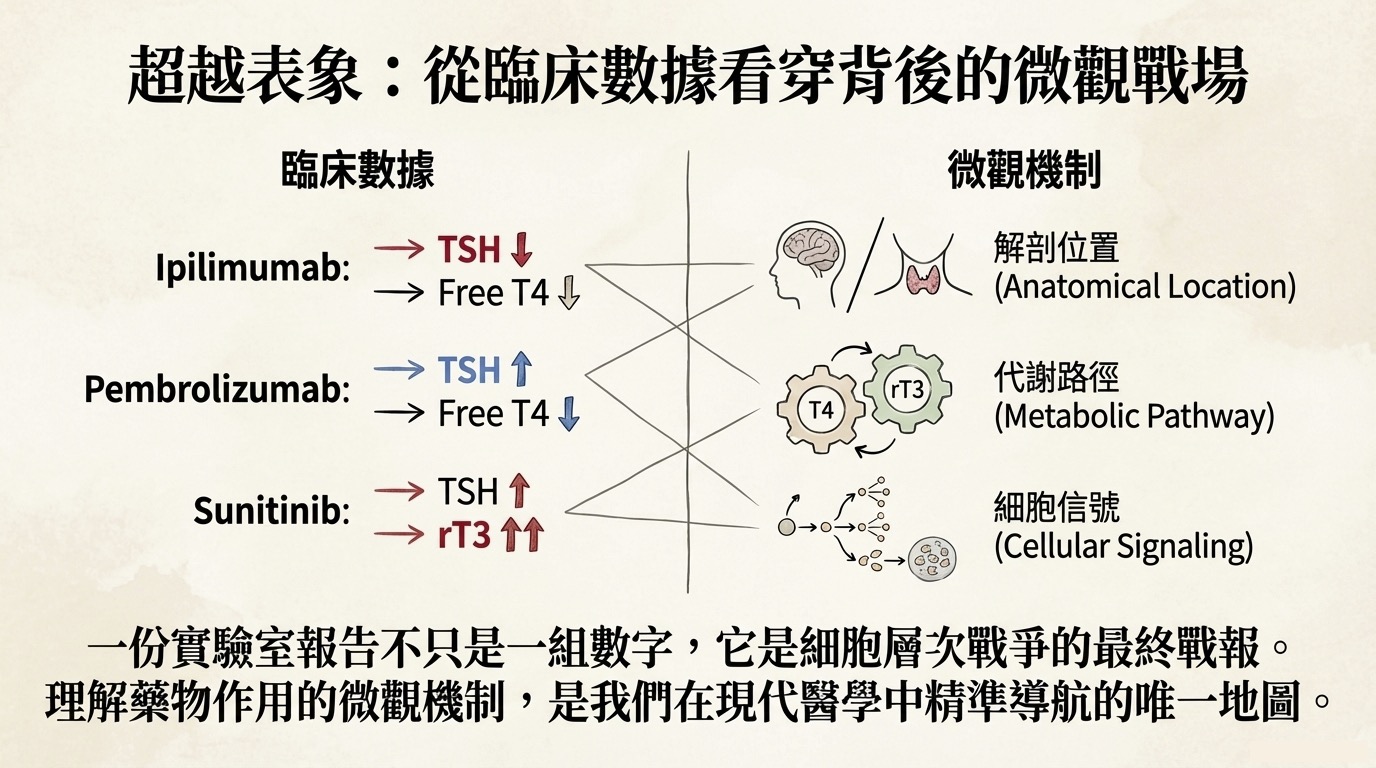
2. 代謝失調:消耗性甲狀腺低下 (Consumptive Hypothyroidism)
這是臨床上容易被忽視的機制。
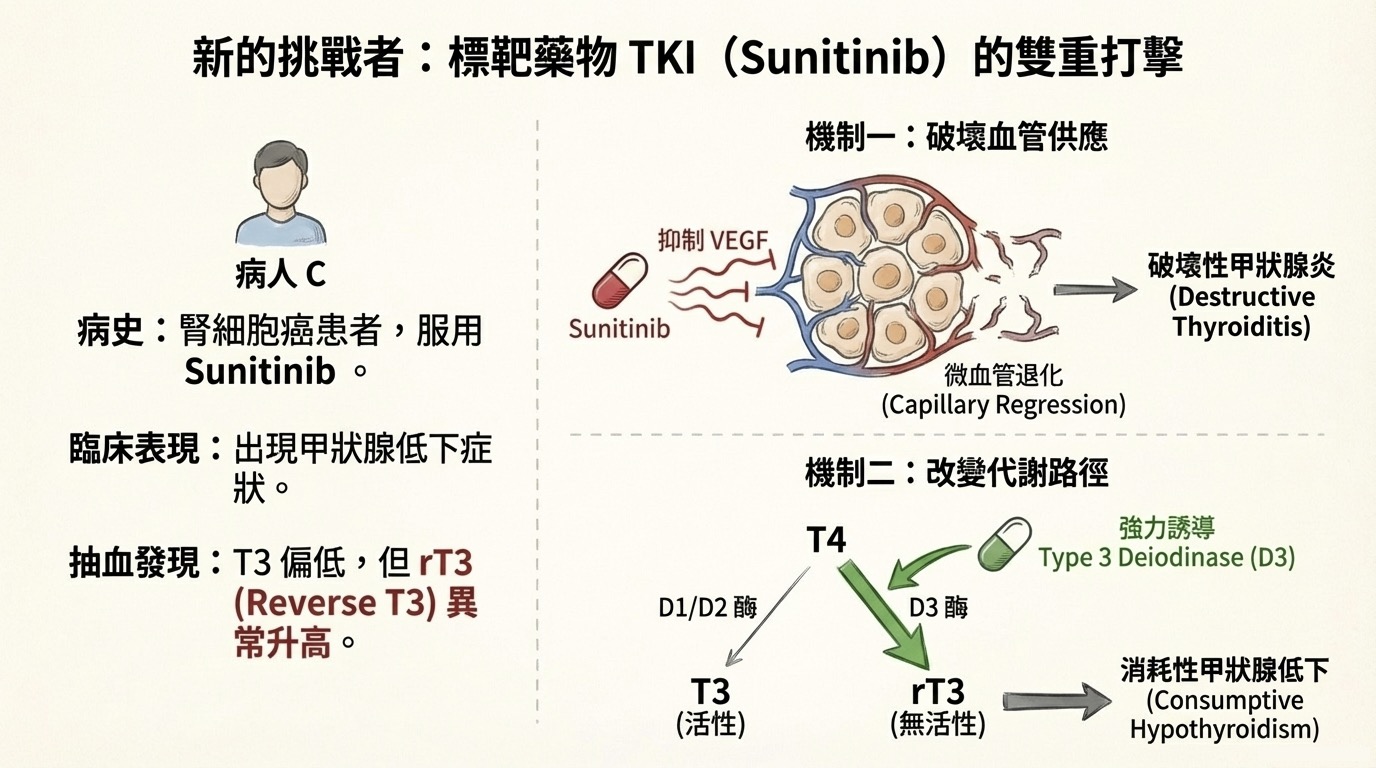
- 逆向適應 (Maladaptation): 當甲狀腺組織缺氧 (Hypoxia) 時,會誘導 HIF-1,進而大量表現 Type 3 Deiodinase (D3)。
- D3 的角色: D3 是甲狀腺素的「滅活酵素 (Inactivator)」。它會將活性的 T4 轉化為完全無活性的 rT3 (Reverse T3)。
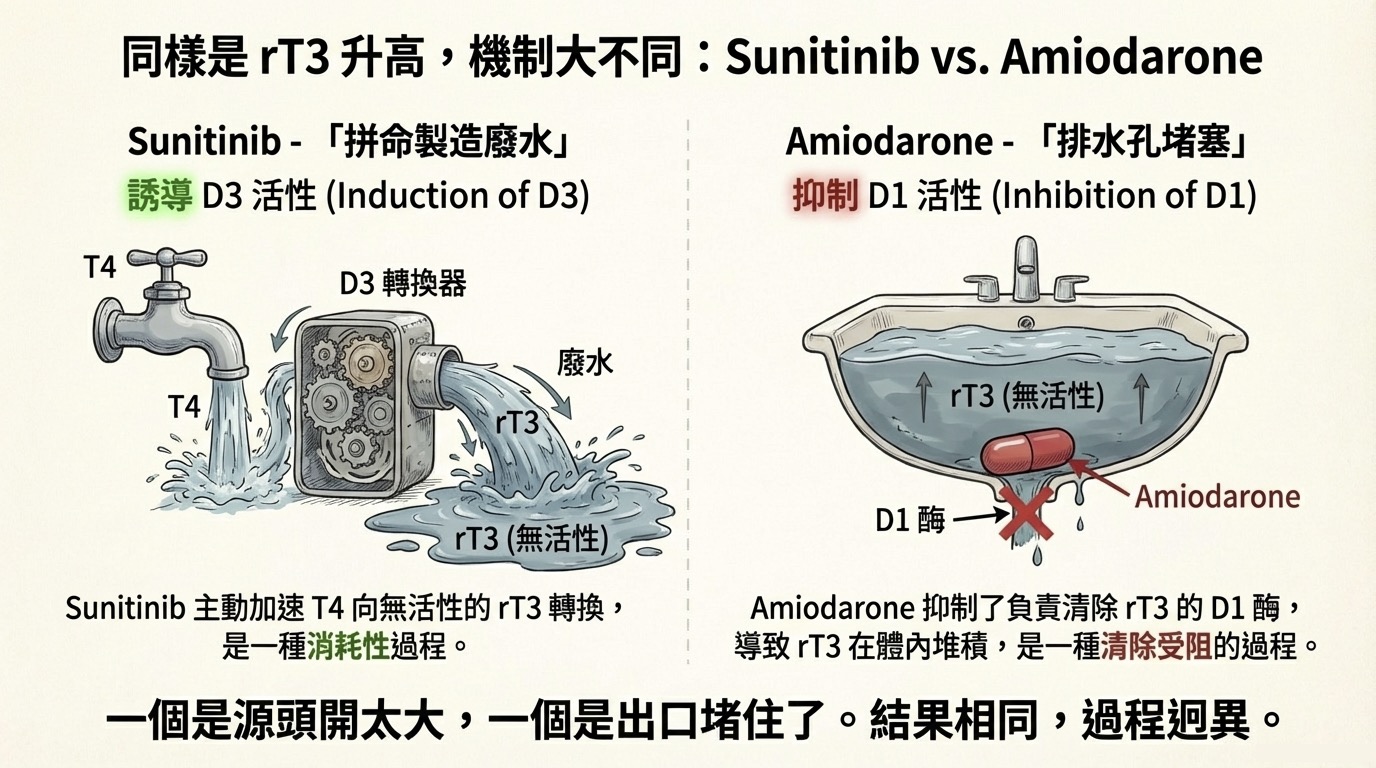
- 臨床後果: 這就像把水龍頭開到最大拼命製造廢水。體內的 T4 和 T3 被快速消耗,導致即使甲狀腺還能製造,也趕不上消耗的速度。
- 抽血特徵: rT3 異常升高,同時 T3 低下。
- 與 Amiodarone 的區別: Amiodarone 導致 rT3 高是因為「排水孔塞住(抑制 D1 清除)」;Sunitinib 是因為「瘋狂製造廢水(誘導 D3 生成)」。
第二部分:免疫檢查點抑制劑 (ICI) 的標靶誤傷
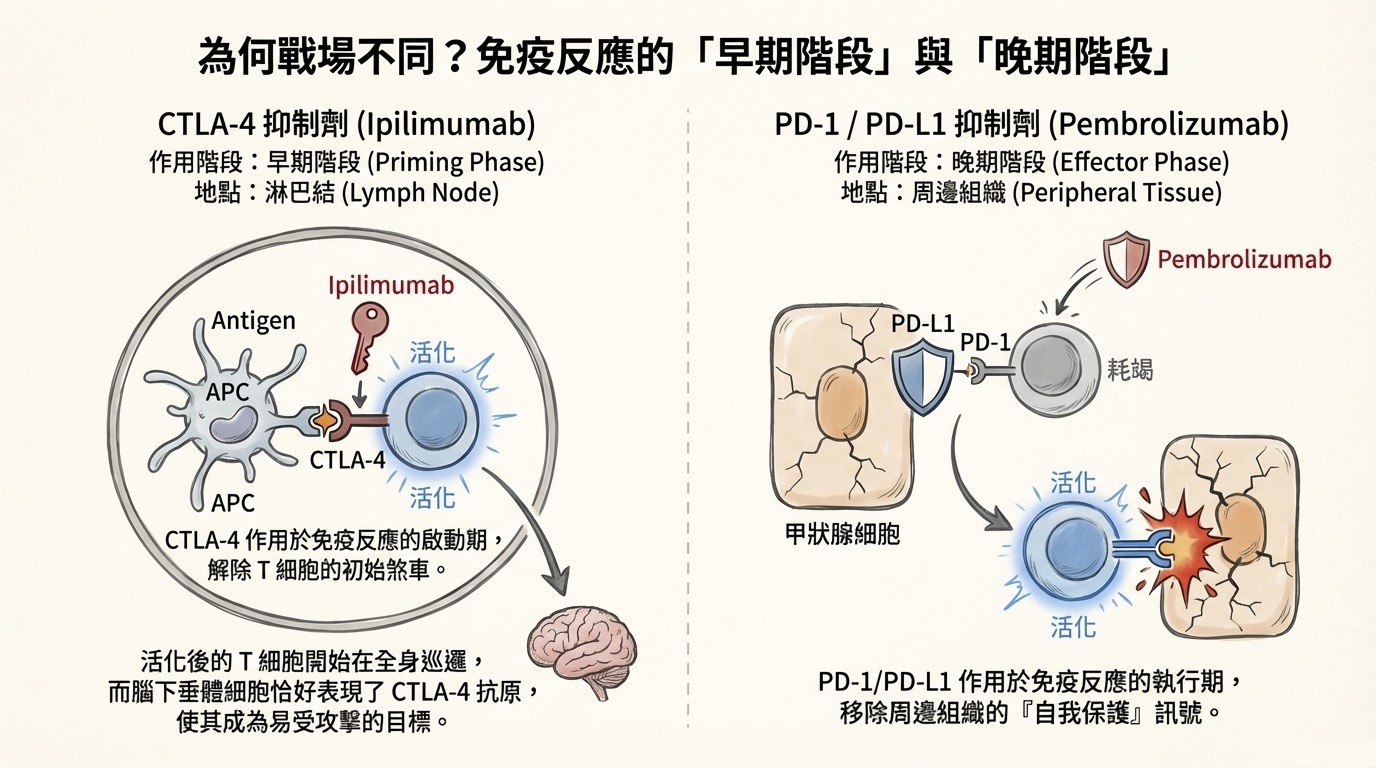
概念: 這些藥物旨在「放開免疫系統的煞車」以攻擊癌細胞,但煞車一鬆,免疫系統便可能攻擊自家的內分泌腺體。 臨床上必須嚴格區分 CTLA-4 與 PD-1 抑制劑,因為它們攻擊的「解剖位置」截然不同。
1. CTLA-4 Inhibitors (如 Ipilimumab)
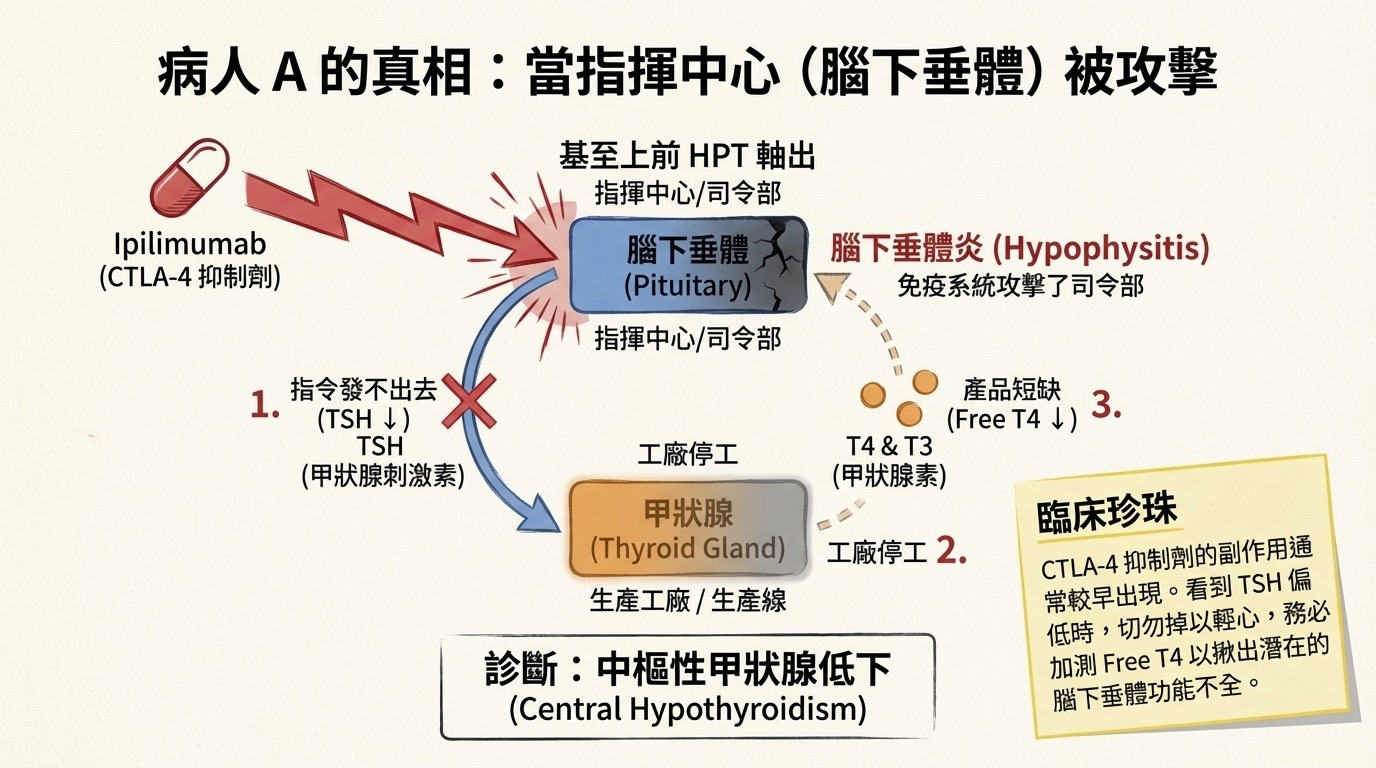
- 攻擊熱點: 腦下垂體 (Pituitary)。
- 微觀機制 (Priming Phase): CTLA-4 作用於免疫反應的早期(淋巴結中)。腦下垂體細胞表面剛好異位表現 (Ectopic expression) CTLA-4 抗原。當藥物在淋巴結解除了 T 細胞的武裝限制,這些 T 細胞出巡時,第一眼就鎖定腦下垂體進行攻擊。
- 病變: 腦下垂體炎 (Hypophysitis)。
- 臨床表現: 病人常先出現頭痛、視野模糊。
- 荷爾蒙特徵: 中樞性甲狀腺低下 (Central Hypothyroidism)。
- TSH 低 (腦下垂體壞了,無法發號施令)。
- Free T4 低 (下游工廠沒收到訂單,停止生產)。
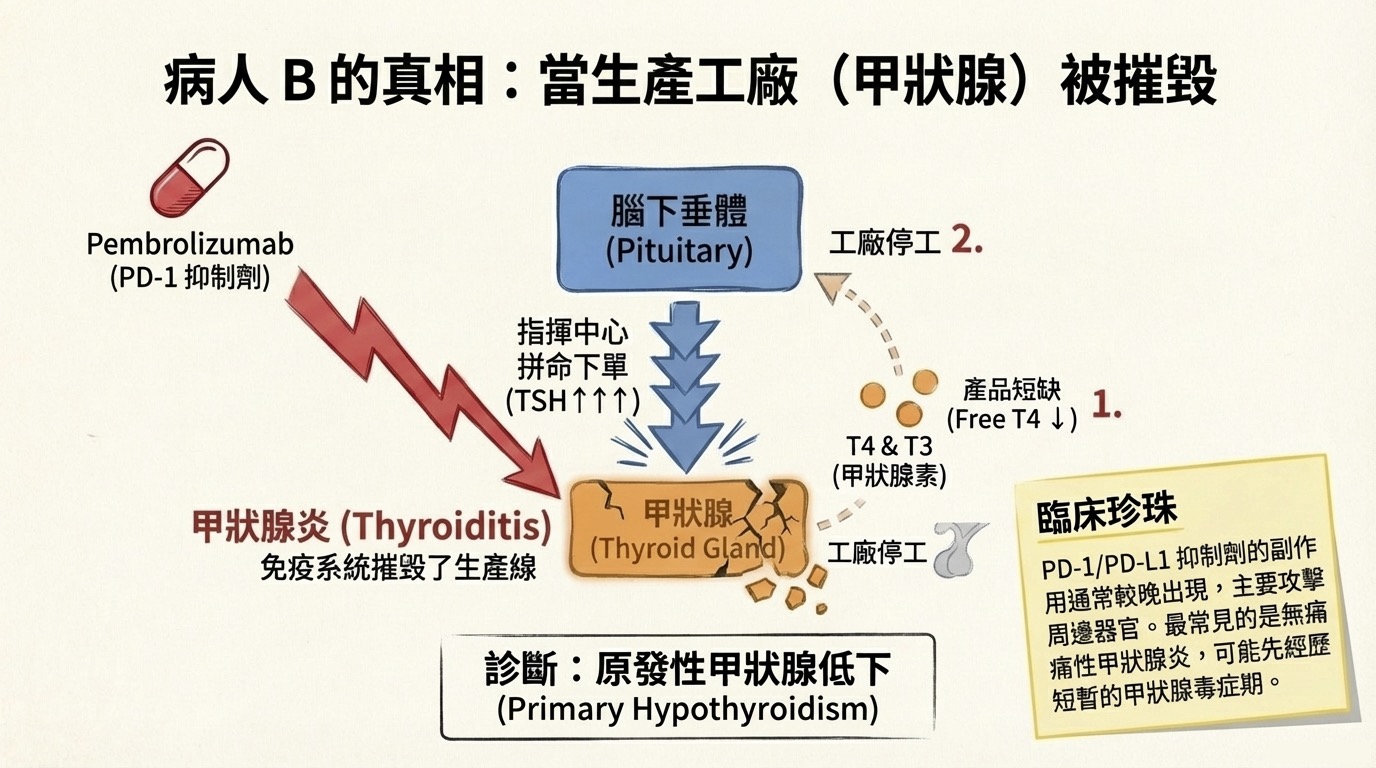
2. PD-1 / PD-L1 Inhibitors (如 Pembrolizumab, Nivolumab)
- 攻擊熱點: 甲狀腺 (Thyroid) 本身。
- 微觀機制 (Effector Phase): PD-L1 表現於周邊組織(含甲狀腺)以作為「免戰牌」。當藥物撕下這張保護膜,原本潛伏在組織周圍的 T 細胞便直接攻擊甲狀腺濾泡。
- 病變: 無痛性甲狀腺炎 (Painless Thyroiditis) 或破壞性甲狀腺炎。
- 荷爾蒙特徵: 原發性甲狀腺低下 (Primary Hypothyroidism)。
- Free T4 低 (工廠被炸毀)。
- TSH 高 (腦下垂體正常,拼命喊話要求生產)。
【臨床實戰演練:疲倦的癌症病人】
情境: 兩位癌症病人皆主訴嚴重疲倦與怕冷,Free T4 皆為 0.6 ng/dL (低)。
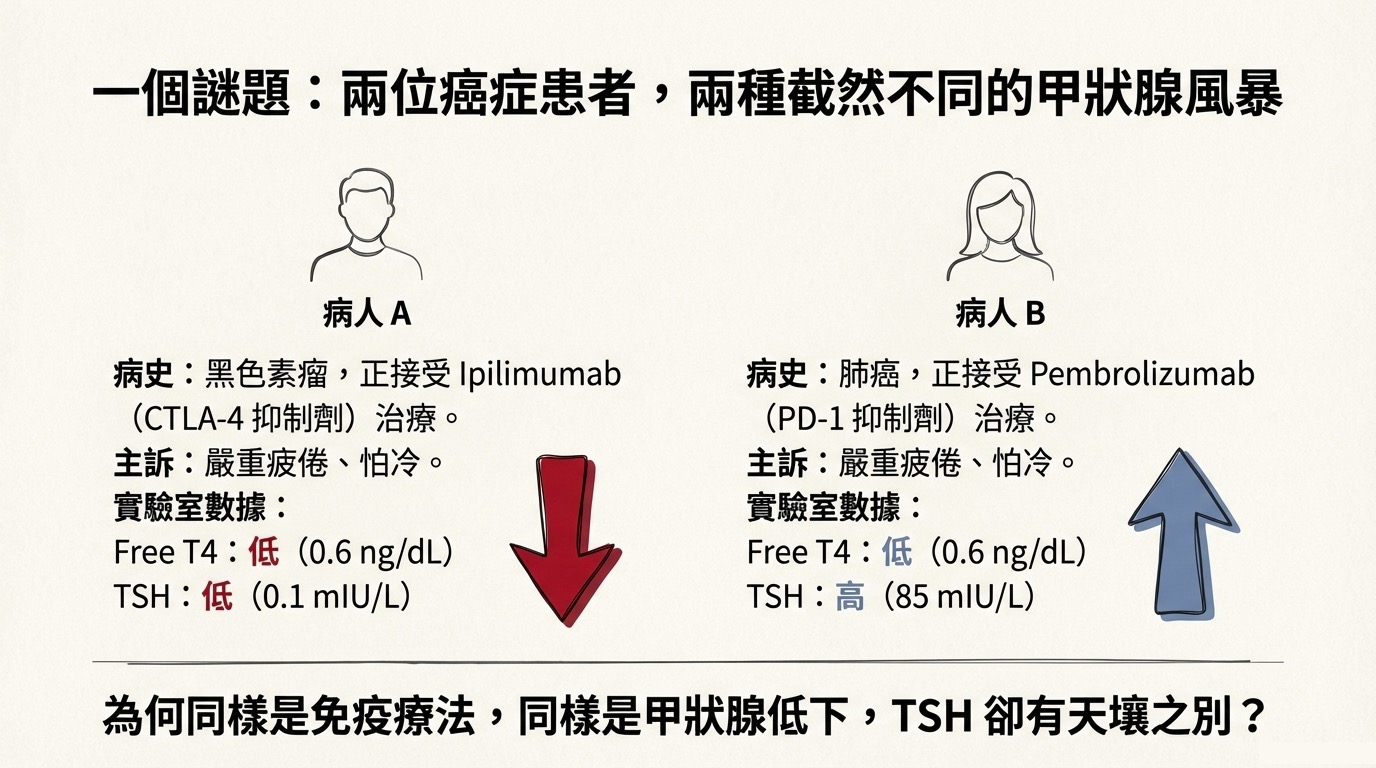
- 病人 A (Ipilimumab): 驗出 TSH 0.1 mIU/L (低) 診斷為 Hypophysitis (中樞性低下)。
- Pearl: 若只看 TSH 偏低常被誤認為沒事,必須加驗 Free T4 才能確診腦下垂體損傷。
- 病人 B (Pembrolizumab): 驗出 TSH 85 mIU/L (高) 診斷為 Thyroiditis (原發性低下)。
第三部分:免疫重建與 Alemtuzumab
藥物: Alemtuzumab (Anti-CD52),用於多發性硬化症 (MS)。
- 獨特副作用: Graves' disease (葛瑞夫茲氏病)。
- 機制: 這是一種 免疫重建症候群 (Immune Reconstitution)。藥物會先將免疫細胞「清零」。當免疫系統重新恢復時,新生出的 B 細胞可能會出現調節失控,產生自體抗體 (TRAb) 攻擊甲狀腺,導致甲狀腺亢進。
第四部分:微觀病理 (Advanced Micro-mechanisms)
這裡深入探討幾個臨床上容易混淆,但機制上有根本差異的現象。
1. Amiodarone 的細胞層次戰爭:Type 1 vs. Type 2
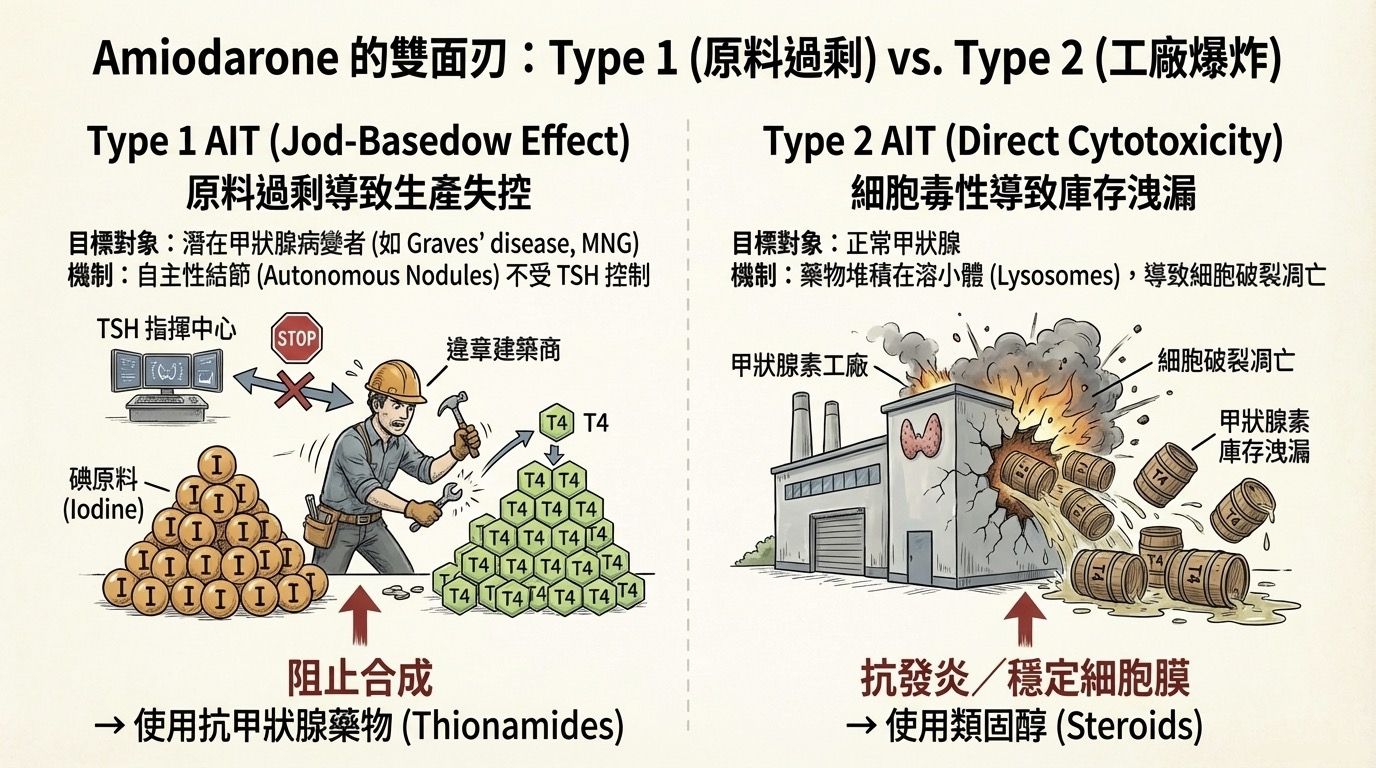
同樣是 Amiodarone 造成的甲狀腺毒症,為什麼有人是 Type 1,有人是 Type 2?這不是隨機的。
- Type 1 (Jod-Basedow Effect):
- 基礎: 病人原本就有潛在病變(如潛伏的 Graves' 或結節)。這些區域具有**「自主性 (Autonomy)」**。
- 機制: 它們不受 TSH 控制。當 Amiodarone 帶來海量碘原料,這些自主區域如同拿到免費建材的違章建築商,瘋狂製造 T4/T3。
- 治療暗示: 需用抗甲狀腺藥物(阻止合成)。
- Type 2 (Direct Cytotoxicity):
- 基礎: 發生在正常甲狀腺上。
- 機制: 藥物代謝物堆積在濾泡細胞的 溶小體 (Lysosomes),導致磷脂質代謝異常 (Phospholipidosis),最終引起細胞破裂。這是「工廠爆炸」,庫存傾洩而出。
- 治療暗示: 需用類固醇(抗發炎/穩定細胞膜)。
2. Lithium 的隱藏手段:Wnt/-catenin 通路
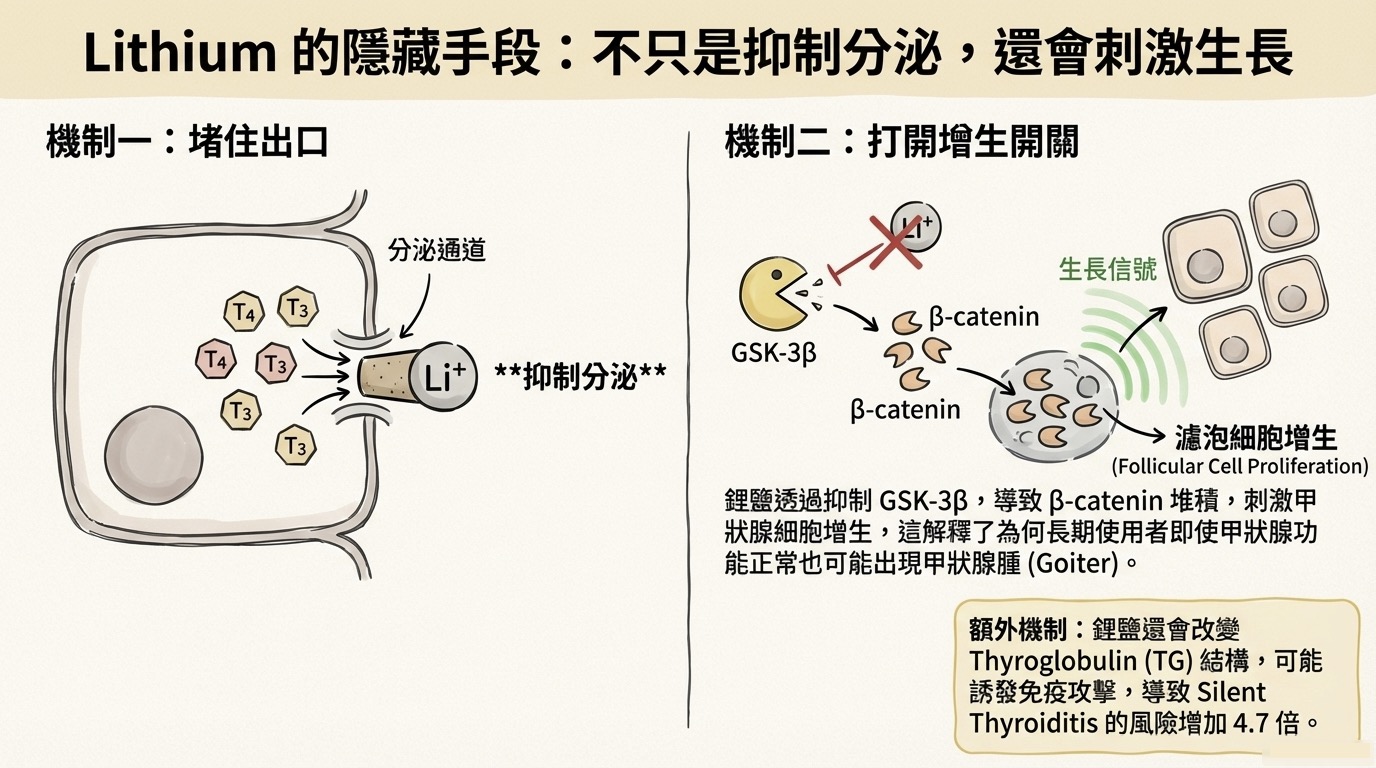
為何長期服用鋰鹽者,即便 TSH 正常,仍可能出現甲狀腺腫 (Goiter)?
- 傳統機制: 抑制分泌 TSH 升高 腫大(這只能解釋部分)。
- 分子機制: 鋰鹽抑制了 GSK-3。GSK-3 原本負責分解 -catenin。被抑制後,-catenin 堆積,這在細胞內模擬出一種「生長訊號」,直接刺激濾泡細胞增生。
- 結構缺陷: 鋰鹽還會改變 Thyroglobulin (TG) 的立體結構,產出有缺陷的 TG,這容易誘發免疫攻擊,解釋了為何鋰鹽使用者發生 Silent Thyroiditis 的機率是常人的 4.7 倍。
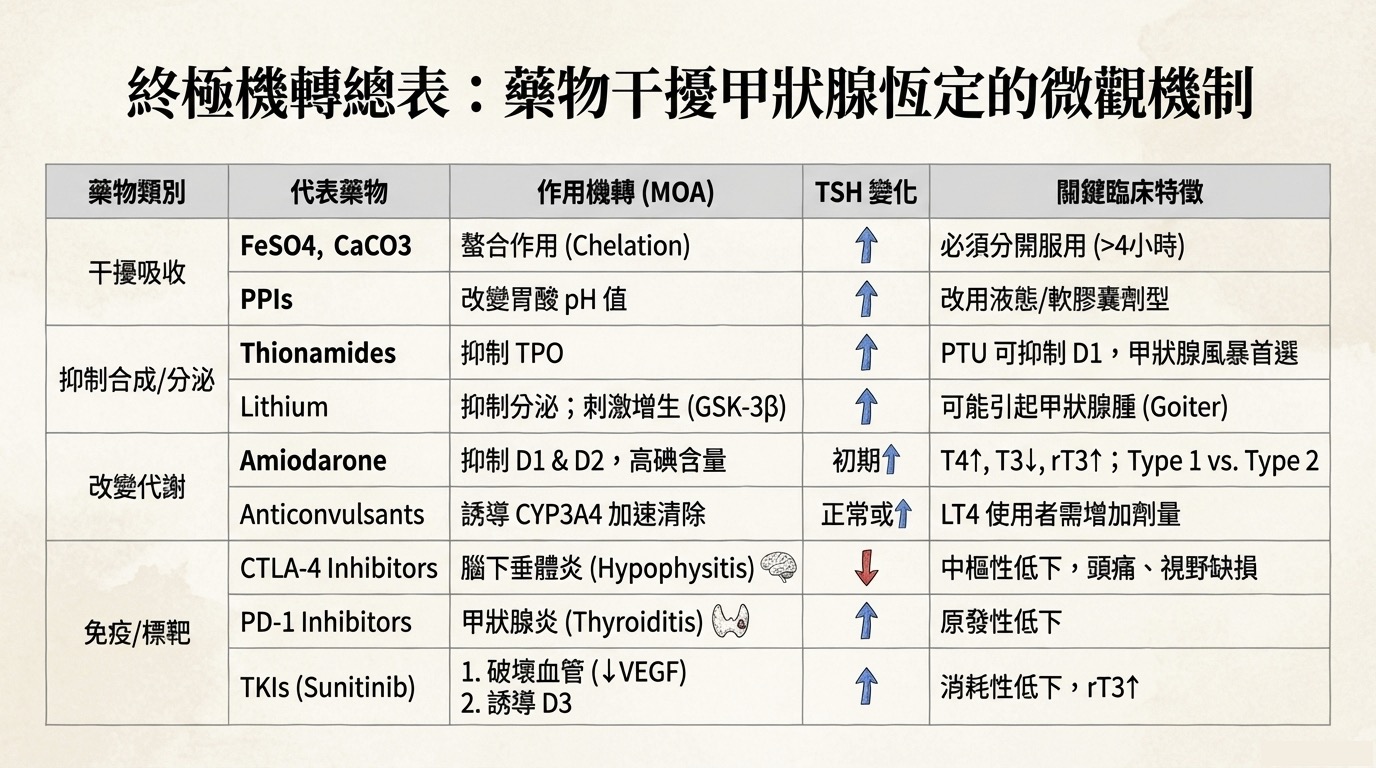
🎓 課程總結:甲狀腺藥物效應終極表